Los métodos computacionales dedicados al estudio de moléculas y materia condensada son de vital importancia en física, química y ciencia de materiales. Esta metodología es utilizada para describir mecanismos a nivel atómico y acelerar el diseño de materiales. A pesar de la gran variedad de metodologías computacionales, el cálculo de la estructura de electrónica es el cuello de botella que limita la velocidad de cómputo y escalabilidad.
El aprendizaje automático o “machine learning” (ML) es un subconjunto de la inteligencia artificial que permite a las computadoras aprender de los datos y mejorar su rendimiento por medio de algoritmos capaces de analizar una gran cantidad de datos, identificar patrones y realizar predicciones. Por ello, ML es una herramienta poderosa para la predicción de propiedades a nivel atómico con un menor gasto de tiempo y costo computacional en comparación con los métodos convencionales.
En los últimos años, el ML ha sido aplicado en simulaciones moleculares usando como base de aprendizaje los cálculos basados en la teoría del funcional de la densidad (DFT). Sin embargo, DFT induce un error sistemático en el cálculo de las propiedades electrónicas del sistema, lo cual conlleva a la pérdida de precisión.
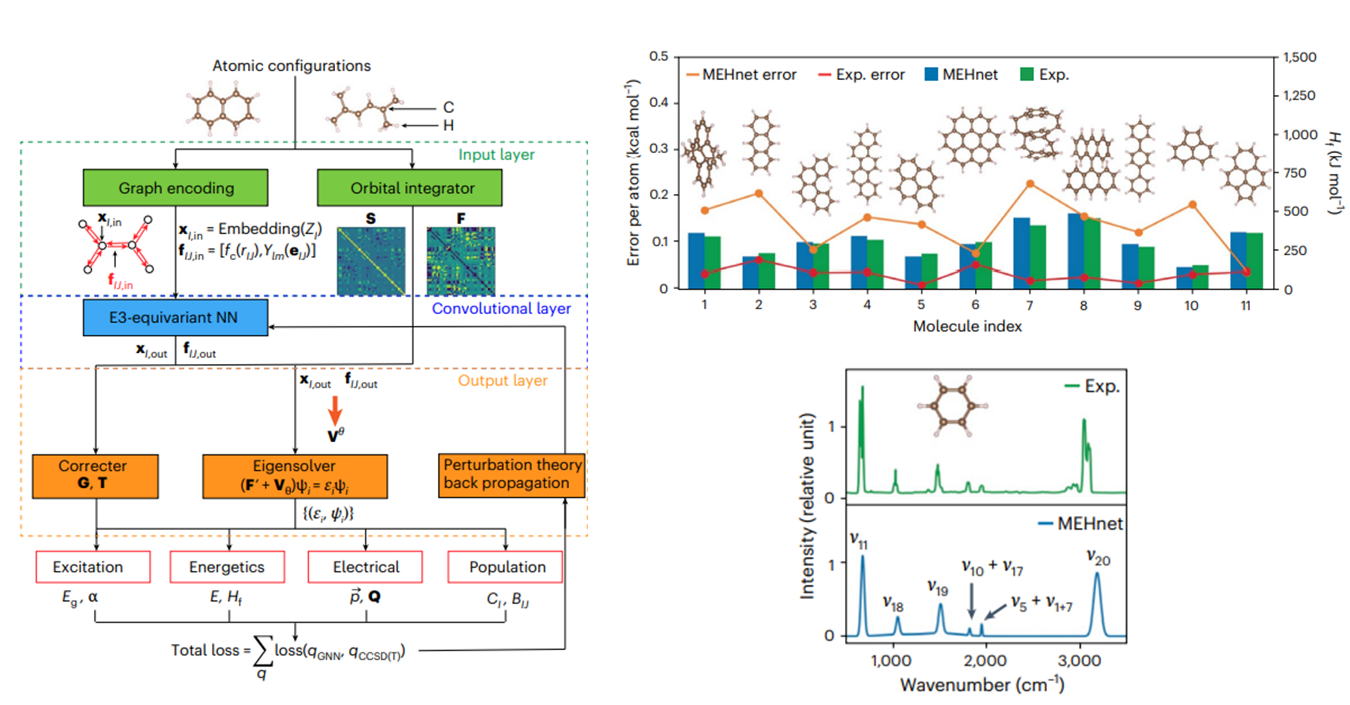
Para solventar esta problemática, científicos del Massachusetts Institute of Technology (MIT) desarrollaron el método de ML-unificado para el cálculo de la estructura electrónica de moléculas orgánicas. Utilizaron la teoría CCSD(T) (del inglés coupled-cluster singles, doubles and perturbative triples), más precisa que DFT, pero que requiere mayor gasto computacional conforme el tamaño de la molécula aumenta. Para entrenar dicho algoritmo, utilizaron 70 moléculas diferentes con 7440 configuraciones atómicas distintas. Entre los resultados que obtuvieron mediante ML, se calcularon entalpias de formación de las moléculas que se compararon con resultados experimentales, encontrando diferencias de 0.1-0.2 Kcal/mol. Asimismo, los espectros de infrarrojo simulados concordaron con datos experimentales en la posición del pico y la intensidad.
A pesar de los buenos resultados, esta metodología aún no ha sido implementada en sistemas periódicos (cristales); sin embargo, los autores creen que esto es posible y se espera que en los próximos años revolucione la forma en que se diseñan los materiales.
Mas información en: nature computational science